Remedi Lab
The major focus of the Remedi laboratory is to study in vivo physiology in various mouse models of diabetes to unravel the underlying mechanisms of pancreatic β-cell failure in glucotoxic stages, and their consequences in both pancreatic and extra-pancreatic tissues. Development of secondary loss of β-cell mass and antidiabetic drug sensitivity in long-standing diabetic patients is not completely understood. Recently, much attention has been gathered on cell plasticity challenging the current paradigms of how diabetes progresses.

The Bionic Mouse

Mice lacking the glycolytic enzyme glucokinase in the pancreas normally never live beyond a few days. The white mouse in the picture lacks glucokinase, but also lacks potassium channels. For this reason it bypasses the need for glucose metabolism in insulin secretion. It remains small and unhealthy but -critically- it can survive (Remedi et al. 2005 Diabetes 54, 2925–2931).
Neonatal Diabetes Mellitus is Genetic and Inherited
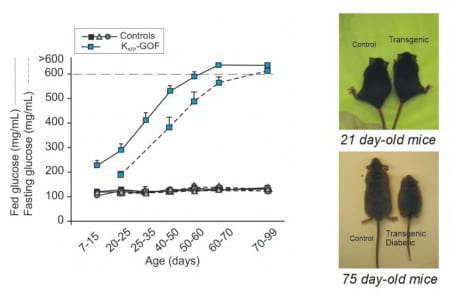
A genetically modified mouse model of Neonatal Diabetes Mellitus reiterated the main features of the human disease: increased blood glucose with development of severe diabetes induced by lack of insulin secretion in response to glucose. As diabetes progresses, these mice show reduction in body weight, among other whole body abnormalities (Remedi et al. 2009 Cell Metabolism, 9, 140-151).
Diabetic Glucotoxicity
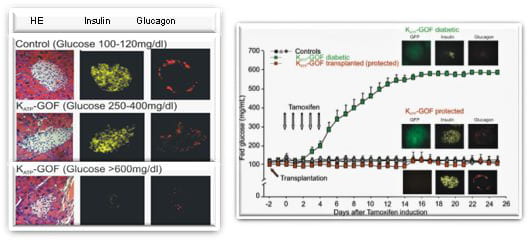
Islets from normal mouse pancreas contain lots of insulin that is released to maintain blood glucose at normal levels. Mice –and people– that express a genetic mutation in potassium channel suffer Neonatal Diabetes Mellitus. The mouse model reveals complex secondary changes, including unexpected disappearance of insulin and glucagon from the islets. Importantly, these consequences are prevented by maintenance of normal blood glucose by either islet transplantation or chronic antidiabetic sulfonylurea therapy (Remedi et al. 2009 Cell Metabolism, 9, 140-151).
Loss of β-cell Identity: to be or not to be? A Reversible Process
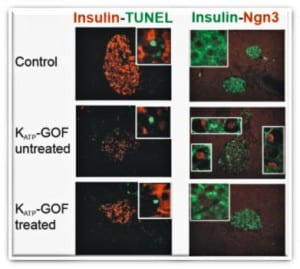
The marked reduction of insulin-containing β-cells in severely diabetic mice (untreated) is not due to increased cell death (TUNEL), but instead to β-cell dedifferentiation to islet progenitor cells (Neurogenin3, Ngn3 positive red nuclei). Strikingly, this process is reversible with the same dedifferentiated cells re-differentiating to mature insulin-containing β-cells following normalization of blood glucose by intensive insulin therapy (treated) (Wang et al. 2014, Cell Metabolism 19:872-882), challenging the paradigm of permanent β-cell damage in long-standing diabetes.
Antidiabetic Sulfonylureas: Transient vs Permanent Neonatal Diabetes?
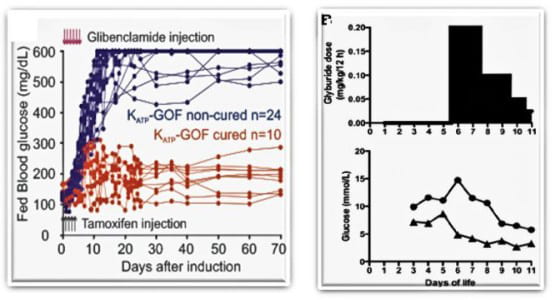
Antidiabetic sulfonylurea therapy at disease onset can cause remission of Neonatal Diabetes in mice (Remedi at al., 2008 PLoS Medicine 5(10):1473-1485; Remedi et al. 2011, Diabetes 60: 2515-22). Strikingly, remission of Neonatal Diabetes has also been shown in patients treated with sulfonylureas very early (Marshall et al. 2015, Diabetes care 38:e38-e39)
The Hyperactive Mouse
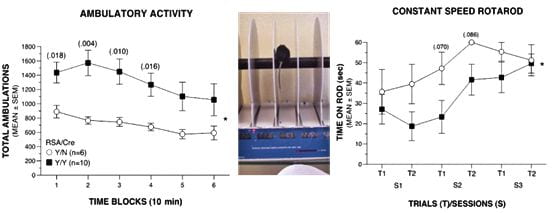
Neurological features in mice expressing mutant channels in the brain. Hyperactivity, impaired motor coordination and balance, decreased muscle strength (unpublished data).
Principal Investigator

Maria S. Remedi, PhD
Professor of Medicine and Cell Biology & Physiology
- Phone: 314-362-6636
- Fax: 314-362-7641
- Email: mremedi@nospam.wustl.edu
Office Location
848 Southwest Tower (Medical campus)
Office Phone (314) 362-6636
Fax (314) 362-7641
Laboratory Location
846 Southwest Tower (Medical campus)
Lab Phone (314) 747-0437
Mailing Address
660 S. Euclid Ave.
Campus Box 8127
St. Louis, MO 63110
Currently seeking graduate students, post-docs and research fellows.






Former Lab Members
Carly Feldman – Undergraduate Research. 2020-2022
Zeenat Asghar Shyr – Postdoctoral Fellow. 2016-2019
Manuela Fortunato – Research Technician. 2017-2018
Christopher Emfinger – PhD student, graduated in 2018
Hannah Conway – Research Technician I, 2016-2019
Alecia Welscher – Research Technician I, 2014-2016
Zhiyu Wang – Clinical Fellow, Washington University
Reka Lorincz – Visiting PhD student, University of Innsbruck – Austria 2017-2018
Stephanie Schiffert NIH Medical Student Summer Research Program, University of Central Florida. 2019
William McAlister- NIH Medical Student Summer Research Program, Brody School of Medicine. 2018
Erin Lindsey – NIH Medical Student Summer Research Program, Saint Louis University. 2017
Amanda Piaruli – NIH Medical Student Summer Research Program, Drexel University. 2016
Undergraduate students
Gabrielle McGinn – Undergraduate student, Washington University. 2019-2020
Matt Fuess – Undergraduate student, Washington University. 2018-2020
Erin Egan – Undergraduate student, Washington University. 2017-2019
Yixi Wang – Undergraduate Student, Washington University. 2016-2018
Arsam Nadeem – Undergraduate Student St. Louis College of Pharmacy. 2016
Leah Yuan – Undergraduate Student, Washington University. 2015-2017
Betsy Abraham- Undergraduate Student St. Louis College of Pharmacy. 2016
Nihar Shah –Undergraduate student, Washington University. 2015-2016
Jonathan Friedman – Undergraduate student, Washington University. 2012-2014
Summer Students
Bailee Rasmussen – Undergraduate summer student, Utah State University. 2019
Eric Hilker – Undergraduate summer student, Truman University 2016
Mariana Alisio – Undergraduate summer student – Washington University, 2015
Hannah Conway – Undergraduate summer student, Loyola University, 2015
Nathan York – Undergraduate summer student, University of Missouri, 2011, 2012
Felesha Clake – High School student, St Louis, 2015